 An Aspergillus fumigatus colony. An Aspergillus fumigatus colony. In the age of next generation sequencing, research on antimicrobial resistance evolution can be done practically using whole genome sequencing (WGS). From the identification of novel resistance mechanisms to spatially tracking the evolution and transmission of resistance alleles, the whole genome approach is fast becoming an invaluable tool in the fight against the antimicrobial resistance crisis. Our goal is to use WGS - by sequencing and analysing ~400 Aspergillus fumigatus genomes from across the globe - to help us research the evolution of antifungal resistance. Antifungal resistance in A. fumigatus, which is fast becoming a credible threat to human health worldwide, is thought to have evolved in both the clinical and environmental setting. This is due to the nature of A. fumigatus; an environmental saprophyte but also a serious fungal pathogen to immunocompromised hosts, where it causes a range of symptoms from mild allergic reactions to invasive aspergillosis of the lungs. Azole antifungals are one of a few classes of drugs available for the treatment and prophylaxis of aspergillosis. Predictably however, after extensive use, resistance to azoles can evolve in A. fumigatus colonising the lungs, and strains with elevated minimum inhibitory concentrations (MIC) are often isolated from sputum samples of infected hosts undergoing azole antifungal therapy. Interestingly though, resistance strains of A. fumigatus have also been isolated from azole naive hosts, which has raised questions as to how and why azole resistance has evolved on these occasions. 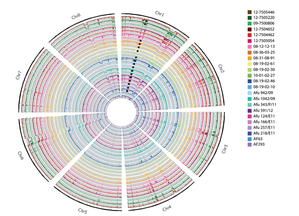 A. fumigatus whole genome alignment - Abdolrasouli et al 2015, mBio. A. fumigatus whole genome alignment - Abdolrasouli et al 2015, mBio. One leading theory suggests that resistance has evolved in the environment under the strong selection of azole fungicide use, which is often applied to agricultural and horticultural crops for the control of plant pathogenic fungi. There are currently two principal "environmental" mutations known to confer resistance to azole antifungals (TR34/L98H and TR46/Y121F/T289A). The two sets of mutations are found in cyp51A, the target of azole antifungals. They are comprised of a tandem repeat (TR) in the promoter region, that increases cyp51A expression, and at least one amino acid substitution that physically alters protein confirmation and decreases azole binding affinity. It is thought that normal dispersal of conidia via asexual reproduction has disseminated these resistance alleles into the wider population, with some resistance conidia inhaled into the lungs of immunocompromised hosts where a resistance phenotype is observed (despite no previous history of azole prescription). 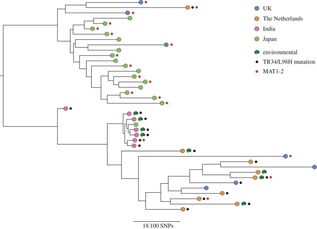 Phylogenetic analysis of A. fumigatus isolates - Abdolrasouli et al 2015, mBio. Phylogenetic analysis of A. fumigatus isolates - Abdolrasouli et al 2015, mBio. To test this theory, our lab applied the WGS approach; we used 24 genome sequences to infer the population structure of resistant and susceptible A. fumigatus isolates collected from both environmental and clinical settings worldwide. This preliminary study showed that sequencing resolution was high enough to identify isolates with and without the TR34/L98H mutation. It identified two distinct clades, which supports previous studies that described this level of structuring. And it showed that A. fumigatus is panmictic and recominogenic, and is probably able to adapt quickly to novel environmental conditions. Our aim now is to build on this previous study and include hundreds more genome sequences to better identify A. fumigatus population structure and evolutionary history in relation to the TR34/L98H and TR46/Y121F/T289A mutations. Hopefully this will enable us to provide a more concrete theory of how, why and when these mechanisms of azole resistance evolved.
Work is well underway... please check back regularly for updates on our progress over the next few years!
0 Comments
Leave a Reply. |
Fisher Lab
|

